Metal and
Oxidative Stress in Yeast
Metals
are of the most important environmental toxics that cause acute and
chronic adverse health effects including cancer. In the last years we
have been focused on the mechanisms used by eukaryotic cells to overcome
metal toxicity. We are particularly interested in the general
aspects of stress response in Saccharomyces cerevisiae
orchestrated by different transcription factors that trigger the reprogramming
of gene expression in response either to toxic metal or to imbalance
homeostasis of essential metals.
Metals are of the most important environmental toxics that cause acute
and chronic adverse health effects including cancer; among these arsenic
compounds are considered the greatest single-cause of ill-health in
the world. Indeed, prenatal arsenic exposure has been found to cause
genome-wide changes in human newborns.The ubiquity of arsenic in the
environment allowed the evolution of very similar defense mechanisms
in organisms ranging from bacteria to man. Indeed, from 20 specific
As(V)-sensitive mutants in mitochondrial genes found, 13 genes have
orthologs in humans. Furthermore, the genome-wide set of S. cerevisiae
deletion strains provided the opportunity to understand the mechanisms
by which arsenic trioxide selectively kills acute promyelocytic leukemia
cells.
 |
Arsenic
(As) compounds are considered the greatest single-cause of ill-health
in the world. Indeed, prenatal arsenic exposure has been found to cause
genome-wide changes in human newborns. The ubiquity of arsenic in the
environment allowed the evolution of very similar defense mechanisms
in organisms ranging from bacteria to man. Thirteen out of the twenty
yeast mitochondrial genes previously involved in As (V) toxicity were
shown to have orthologs in humans. As such S. cerevisiae deletion
strains are powerful tools to understand the mechanisms by which arsenic
trioxide selectively kills acute promyelocytic leukemia cells. Within
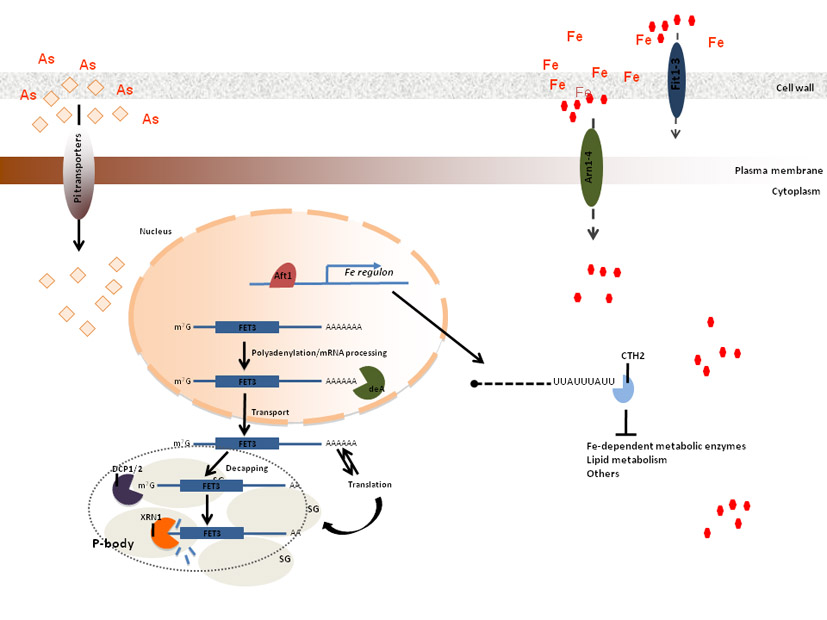
this frame, we have recently shown that Arsenic compounds disturb iron
homeostasis in yeast as well as in mammalian cells (figure above). This
finding can be relevant to future clinical applications. Yap8, one of
the eight members of the Yap family of transcription factors, is a key
regulator in yeast of the expression of two genes encoding the arsenate-reductase
Acr2 and the plasma membrane arsenite efflux transporter Acr3. These
two genes form the Yap8 regulon and we have recently addressed the study
of its very restricted DNA binding specificity using a model of Yap8-DNA
interaction and in vivo and in vitro experimental
approaches. Acr2 and Acr3 constitute the main Arsenics detoxification
pathway. Yap1, the master regulator of the cell antioxidant defenses,
contributes to arsenic stress responses by regulating the expression
of a vacuolar arsenite detoxification pathway encoded by YCF1, although
we have recently shown that the most important role of Yap1 in arsenic
adaptation is through the maintenance of the redox homeostasis disturbed
by inorganic arsenic compounds. To analyze whether Yap1 and Yap8 use
similar mechanisms to transduce the stress signals to the basal transcription
machinery, we are addressing the effect of mutations in specific subunits
of the tail module of the mediator complex.
Yap1 appears to be in the forefront of cadmium stress response. Yap2
is the family member that shares the highest homology with Yap1, but
very little is known about its functional role. Nevertheless it is known
that, when overexpressed, Yap2 confers resistance to several stress
agents, such as cadmium. Given Yap1 and Yap2 similarities it is widely
accepted that a certain degree of functional overlap exists between
both transcription factors, however it is evident that Yap2 plays unique
roles without interference of the realm of Yap1 function. Current investigations
in our laboratory are aimed at understanding of how Yap1 and Yap2 coordinate
their gene target expression, in order to endow the cell with the ability
to overcome cadmium toxicity.
Cobalt
has a rare occurrence in nature, but may accumulate in cells to toxic
levels. We have shown that Cobalt activates Yap1 that alleviate the
oxidative damage caused by this metal. Yap1 partially regulates cobalt
cellular uptake via the regulation of the high affinity phosphate transporter
Pho84. Transcriptomic analysis revealed that Yap1 is a repressor of
the low affinity iron transport Fet4. Although Fet4 repression by Yap1
has no effect on cobalt uptake we are currently investigating whether
it may be its first line of defense against other toxic metals.
 |
Iron
(Fe) is an essential metal to most forms of life. However, the same
chemical properties that make Fe such a central element for life also
make it a strong pro-oxidant that can generate powerful reactive oxygen
species (ROS) through Fenton type reactions. Unlike humans, but similar
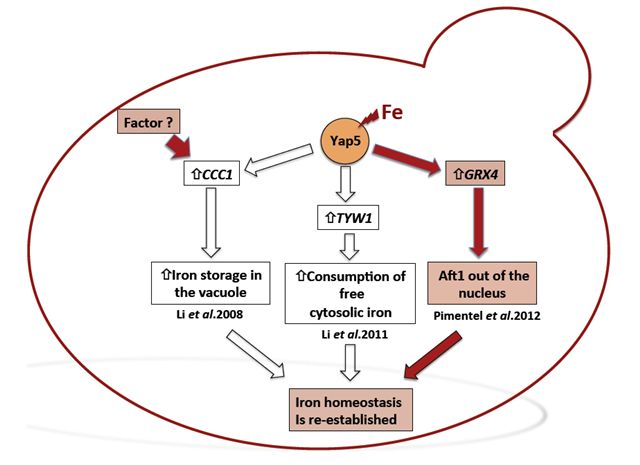
to plants, the yeast cell vacuoles function as iron reservoirs. In yeast,
iron storage is mediated by Ccc1, a vacuolar transporter that effects
the accumulation of iron in the vacuoles. In a high-Fe milieu, CCC1
gene deletion is lethal and its expression is regulated by the transcription
factor Yap5. We have analysed the transcriptional response of the yap5
null mutant subjected to high concentrations of iron, having identified
GRX4 gene as a Yap5 target. This gene encodes a monothiol glutaredoxin
previously known to be involved in the regulation Aft1 nuclear localization.
Consistently we have shown that in the absence of Yap5, Aft1 nuclear
exclusion is slightly impaired.
Overall
our recent studies provide further evidence that cells control metal
homeostasis or metal toxicity by using multiple pathways.
BACK
TO THE TOP
Cross-talk
between different Yap factors
Although the binding
of promoters by multiple transcriptional regulators has been characterized
as a specific feature of higher eukaryotes, many yeast genes were found
in simultaneous association with several transcription factors. Within
this context, Yap4 has been shown to be regulated either by Msn2 under
conditions of osmotic stress or by Msn2 and Yap1 under oxidative stress.
Using computational approaches, transcriptional regulatory networks
were derived that were shown to control several cellular processes such
as the cell cycle, environmental stress, development and metabolism.
It was also predicted
through mathematic modeling that Yap4 interacts with Yap6. Several studies
have shown that the Yap family members appear to have a degree of functional
overlap as well as distinct physiological roles. It was indeed shown
that it is possible, using DNA microarrays, to distinguish between the
functions of Yap1 and Yap2. These two transcription factors although
having overlapping functions, both activate non-overlapping gene sets.
One possible model put
forward to describe the Yap network is that during oxidative stress
the Yap1 and Yap2 homodimers activate distinct regulons whereas Yap1/Yap2
heterodimers collaborate to repress a separate regulon. Similarly, the
yap8yap1 strain has increased metalloid sensitivity than either single
mutant. The expression of the Yap8 target genes ACR2, ACR3 and YCF1
is also significantly regulated by Yap1, both of which have been shown
to bind the YRE variant, TGATTAATAATCA. We have constructed a Yap0 mutant
strain, a strain deleted in all the Yap factors which is a valuable
tool to discriminate overlapping functions as well as the interplay
between them.
BACK
TO THE TOP
Nitrosative Stress
Response in Desulfovibrio gigas
Desulfovibrio
gigas is an anaerobic
microorganism that belongs to the group of sulfate reducing bacteria
(SRB). SRB are metabolic versatile microorganisms widespread in nature
as well as in the human gastrointestinal tract, being often exposed
to reactive nitrogen species (RNS), produced by other bacteria or by
the human innate immune system. To cope with RNS deleterious effects
microorganisms have developed several mechanisms that afford protection
against nitrosative stress. Although Desulfovibrio is the most studied
genus of SRB, the mechanisms and regulatory elements involved in this
protection are still poorly understood. Using D. gigas as a
model organism we addressed the importance of several transcriptional
factors in nitrosative stress response. Our lab was pioneer in constructing
mutants of this bacterium, and we are using this approach to investigate
whether those factors are involved in NO detoxification.
In
Desulfovibrio gigas we have already shown that the Rubredoxin
oxygen oxidoreductase, ROO, affords protection against nitrosative stress.
Our recent findings have shown that ROO gene expression is regulated
by NorR1L, an ortholog of NorR from E.coli. Furthermore we found a NorR1L
paralog (NorR2L) within the genome of D. gigas whose function
under stress is currently being investigated.
Lately, we have
found that D.gigas HcpR, a transcription factor belonging to
the family of CRP/FNR, regulates several genes encoding proteins involved
in nitrite and nitrate metabolism. Accordingly, an hcpR mutant strain
displays a growth sensitivity phenotype to NO, strongly supporting a
relevant role of this factor under nitrosative stress. Moreover, we
found that several Desulfovibrio spp. possess HcpR paralogs
bringing to light the possibility that these species may exhibit higher
tolerance to nitrosative stress. Detailed structural and functional
analyses of the sequences need, however, to be evaluated in order to
fully understand their role in stress response. Further work is in progress
in order to clarify the role of HcpR paralog in D.gigas.
Within
the context of dissecting the mechanisms of nitrosative stress adaptation
in SRBs, we aim at determining the transcription profile of D.gigas
Hcpr, NorR and their respective paralog genes null mutants, after
challenge with NO.
Desulfovibrio
gigas Genome: past and future
Together
with STAB
VIDA Lda.,
we have recently sequenced the D. gigas genome providing insights into
the integrated network of energy conserving complexes and structures
present in this bacterium.
The
genome of D. gigas (CP006585) consists of one circular chromosome
of 3,693,899 base-pairs (bp) having 3,370 genes of which 3,273 are protein
coding classified according to its predicted COG function. The genome
has a G+C content of 63.4% that reflects a biased codon usage as such
D. gigas prefers high G+C codons (66.87%), with a clear preference for
cytosine (C) in the 3rd position. The genome is very compact as observed
by its gene density of 1,128 bp per gene and the average length of each
gene is 993 bp. Its genome contains solely 17 genes encoding transposases
and only a single rRNA operon indicating a decreased genome rearrangement,
as multiple rRNA operons serves as sites for homologous recombination.
The plasmid of this bacterium (CP006586) has a size of 101,949bp, containing
75 ORFs, of which 72 are coding regions.
The
size of Desulfovibrio gigas is larger than the one of other
Desulfovibrio spp. Its length is of 5 to 10 µm and the
width of 1.2 to 1.5 µm whereas the other species have a cell size
of 3 to 5 µm by 0.5 to 1 µm. We have found several genes
which can explain the large size of this bacterium
A
survey of the genome of D. gigas for CRISPR repeats, revealed
the presence of 6 CRISPR repeats with two of them being flanked by Cas
operons. The CRISPRs are loci encompassing several short repeats functioning
as an adaptive microbial immune system, that have also been shown to
limit horizontal gene transfer by preventing conjugation and plasmid
transformation.
Gene
duplications were found such as those encoding fumarate reductase, formate
dehydrogenase and superoxide dismutase. Complexes not yet described
within Desulfovibrio genus were identified: Mnh complex, a v-type ATP-synthase
as well as genes encoding the MinCDE system that could be responsible
for the larger size of D. gigas when compared to other members
of the genus. A low number of hydrogenases and the absence of the codh/acs
and pfl genes, both present in D. vulgaris strains, indicate that intermediate
cycling mechanisms may contribute substantially less to the energy gain
in D. gigas compared to other Desulfovibrio spp. This might
be compensated by the presence of other unique genomic arrangements
of complexes such as the Rnf and the Hdr/Flox, or by the presence of
NAD(P)H related complexes, like the Nuo, NfnAB or Mnh.
Structural representation
of the circular chromosome (A) and plasmid (B) of Desulfovibrio
gigas. Circular representations, from inside to the outside represent:
(i) GC skew, richness of guanine over cytosine in the positive strand
represented in green and cytosine over guanine represented in red; (ii)
GC content, below average in purple, above average in gold; (iii) positive
strand coding regions (below) and negative strand coding regions (above)
colored according to COG functional terms of the best hit obtained from
Blastp program; (iv) nucleotide position indicated in circular scale.
Taking
advantage of the complete genome sequence is our future goal to have
a general picture of the transcriptome of
Desulfovibrio gigas subjected to different
stress conditions that may interfere with its metabolism
BACK
TO THE TOP




