



A Nobel for the computational tools that help us unveil the mysteries of proteins
Oeiras, 07 October 2024
The 2024 Nobel Prize in Chemistry was awarded to researchers David Baker, Demis Hassabis and John Jumper for their work in computational protein design and protein structure prediction, which is enabling the understanding of many biological processes at the microscopic level and leading to major developments in biotechnology and health.
A Nobel for the computational tools that help us unveil the mysteries of proteins
Diana Lousa and Manuel M. Melo
Creating new proteins to treat diseases such as cancer, defend ourselves against viruses and bacteria or degrade pollutants such as plastic sounds like fiction, but it is now possible. The discoveries that it is possible to use computational tools to create proteins and unravel their structure, awarded this year's Nobel Prize in Chemistry, have revolutionized and exponentially accelerated our ability to respond to these and other problems.
Proteins are long molecules formed from the sequential linking of smaller molecules - amino acids. The chemical diversity of the various amino acids means that proteins tend to fold in a way that is defined by their sequence. This three-dimensional structure that proteins adopt determines their functions. However, even knowing the sequence of amino acids that make up a protein, it is challenging to predict which three-dimensional structure it will end up adopting.
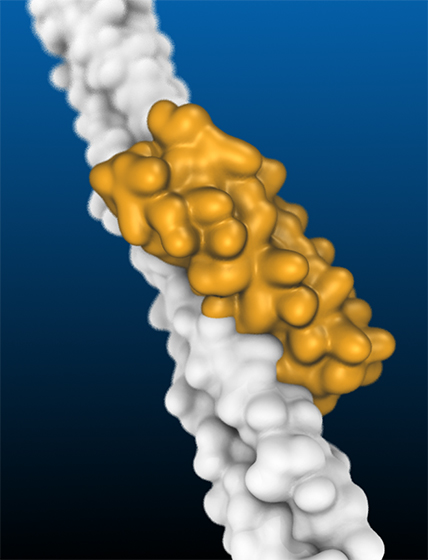 The scientists who won this year’s Nobel Prize for Chemistry, David Baker, Demis Hassabis and John Jumper, were able to solve two major puzzles in biochemistry related to this problem: How can we efficiently and accurately predict the structure of a protein from its amino acid sequence? And how can we design the sequence of a protein so that it has the desired structure and properties?
The scientists who won this year’s Nobel Prize for Chemistry, David Baker, Demis Hassabis and John Jumper, were able to solve two major puzzles in biochemistry related to this problem: How can we efficiently and accurately predict the structure of a protein from its amino acid sequence? And how can we design the sequence of a protein so that it has the desired structure and properties?
To solve these challenges, the laureates used two types of computational strategies. The first, implemented by David Baker, used knowledge of the chemical and physical behavior of amino acids to create computer models capable of relating the sequence and structure of proteins. The second strategy, developed by Demis Hassabis and John Jumper, took advantage of major developments in the field of artificial intelligence to train models to make this prediction. Here, instead of inferring the complexity of protein behavior from the properties of the constituent amino acids, they used several thousand protein structures that had already been determined - very laboriously - by experiment to teach computer models to directly relate sequence to structure, taking advantage of knowledge that can be extracted from multiple sequence alignments.
The models developed have already created hundreds of millions of predictions of protein structures, which make it possible to understand the inner workings of many biological processes at a microscopic level and will lead to major developments in the fields of biotechnology and health. And the models have not only been applied to existing proteins. They also allow us to design new amino acid sequences tailored to the functions we want them to perform. In the international BioPlaTTAR and EvaMobs projects that we coordinate, we design proteins which are optimized to block the surface of viruses in order to prevent them from entering our cells. As we can create specific proteins for different targets, this strategy can be applied to a wide range of diseases and allow us to fight future pandemics.
These are exciting times in the field of computational structural biology!
Diana Lousa (Researcher in the Protein Modelling Lab) and Manuel N. Melo (Lab Head of the Multiscale Modelling Lab)
Figure caption: Illustration of a protein created by ITQB NOVA researchers using computational tools (in yellow) that has been designed to efficiently fit onto a structure on the surface of the SARS-CoV-2 virus (in white). By binding to the surface of the virus, the designed protein prevents it from entering our cells and inhibits infection.


